薬効分類名ヒト型抗ヒトIL-1 β モノクローナル抗体
一般的名称カナキヌマブ(遺伝子組換え)
イラリス皮下注射液150mg
いらりすひかちゅうしゃえき150mg
ILARIS solution for s.c. injection 150mg
製造販売/ノバルティスファーマ株式会社
重大な副作用
その他の副作用
併用注意
抗TNF製剤
重篤な感染症発現のリスクが増大するおそれがある。また、他の抗IL-1製剤と抗TNF製剤との併用により、重篤な感染症の発現頻度増加が認められているため、本剤との併用は行わないことが望ましい。
共に免疫抑制作用を有するため。
1. 警告
- 1.1 本剤投与により、敗血症を含む重篤な感染症等があらわれることがあり、本剤との関連性は明らかではないが、悪性腫瘍の発現も報告されている。本剤が疾病を完治させる薬剤でないことも含め、これらの情報を患者に十分説明し、患者が理解したことを確認した上で、治療上の有益性が危険性を上回ると判断される場合にのみ本剤を投与すること。また、本剤の投与において、重篤な感染症等の副作用により、致命的な経過をたどることがあるので、緊急時に十分に措置できる医療施設及び医師のもとで投与し、本剤投与後に副作用が発現した場合には、速やかに担当医に連絡するよう患者に注意を与えること。[1.2 参照],[2.1 参照],[8.1 参照],[8.2 参照],[8.6 参照],[9.1.1 参照],[9.1.3 参照],[9.1.4 参照],[11.1.1 参照]
- 1.2 敗血症等の致命的な感染症が報告されているため、十分な観察を行うなど感染症の発現に注意すること。[1.1 参照],[2.1 参照],[8.1 参照],[8.2 参照],[9.1.1 参照],[9.1.3 参照],[9.1.4 参照],[11.1.1 参照]
- 1.3 本剤についての十分な知識と適応疾患の治療の知識・経験をもつ医師が使用すること。
2. 禁忌(次の患者には投与しないこと)
- 2.1 重篤な感染症の患者[感染症が悪化するおそれがある。][1.1 参照],[1.2 参照],[8.1 参照],[8.2 参照],[9.1.1 参照],[9.1.3 参照],[9.1.4 参照],[11.1.1 参照]
- 2.2 活動性結核の患者[症状が悪化するおそれがある。][8.3 参照],[9.1.2 参照]
- 2.3 本剤の成分に対し過敏症の既往歴のある患者
5. 効能又は効果に関連する注意
-
**〈シュニッツラー症候群〉
- 5.1 「17. 臨床成績」の項の内容を熟知し、臨床試験に組み入れられた患者の臨床症状及び治療歴を十分に理解した上で、適応患者を選択すること。[17.1.6 参照]
- 〈家族性地中海熱〉
- *〈全身型若年性特発性関節炎及び成人発症スチル病〉
6. 用法及び用量
-
〈クリオピリン関連周期性症候群〉
通常、体重40kg以下の患者にはカナキヌマブ(遺伝子組換え)として1回2mg/kgを、体重40kgを超える患者には1回150mgを8週毎に皮下投与する。
十分な臨床的効果(皮疹及び炎症症状の寛解)がみられない場合には適宜漸増するが、1回最高用量は体重40kg以下の患者では8mg/kg、体重40kgを超える患者では600mgとする。
最高用量まで増量し、8週以内に再燃がみられた場合には、投与間隔を4週間まで短縮できる。
なお、症状に応じて1回投与量の増減を検討すること。 -
〈高IgD症候群(メバロン酸キナーゼ欠損症)〉
通常、体重40kg以下の患者にはカナキヌマブ(遺伝子組換え)として1回2mg/kgを、体重40kgを超える患者には1回150mgを、4週毎に皮下投与する。
十分な臨床的効果がみられない場合には追加投与又は適宜漸増するが、1回最高用量は体重40kg以下の患者では6mg/kg、体重40kgを超える患者では450mgとする。 -
〈TNF受容体関連周期性症候群及び家族性地中海熱〉
通常、体重40kg以下の患者にはカナキヌマブ(遺伝子組換え)として1回2mg/kgを、体重40kgを超える患者には1回150mgを、4週毎に皮下投与する。
十分な臨床的効果がみられない場合には追加投与又は適宜漸増するが、1回最高用量は体重40kg以下の患者では4mg/kg、体重40kgを超える患者では300mgとする。 -
〈シュニッツラー症候群〉
**通常、成人にはカナキヌマブ(遺伝子組換え)として体重40kg以下の患者では1回2mg/kgを、体重40kgを超える患者では1回150mgを8週毎に皮下投与する。
十分な臨床的効果がみられない場合には追加投与又は適宜漸増するが、1回最高用量は体重40kg以下の患者では4mg/kg、体重40kgを超える患者では300mgとする。 -
*〈全身型若年性特発性関節炎及び成人発症スチル病〉
通常、カナキヌマブ(遺伝子組換え)として1回4mg/kgを、4週毎に皮下投与する。1回最高用量は300mgとする。
7. 用法及び用量に関連する注意
- 〈効能共通〉
-
**〈クリオピリン関連周期性症候群、高IgD症候群(メバロン酸キナーゼ欠損症)、TNF受容体関連周期性症候群、シュニッツラー症候群、家族性地中海熱〉
- 7.3 投与は1回2mg/kg又は150mgの低用量から開始し、十分な効果がみられない、もしくは再燃がみられた場合に限り、下図を参考に投与量の増量を行うこと。[17.1.1 参照],[17.1.5 参照],[17.1.6 参照]
- 十分な臨床的効果がみられない場合の漸増方法
-
〈クリオピリン関連周期性症候群〉
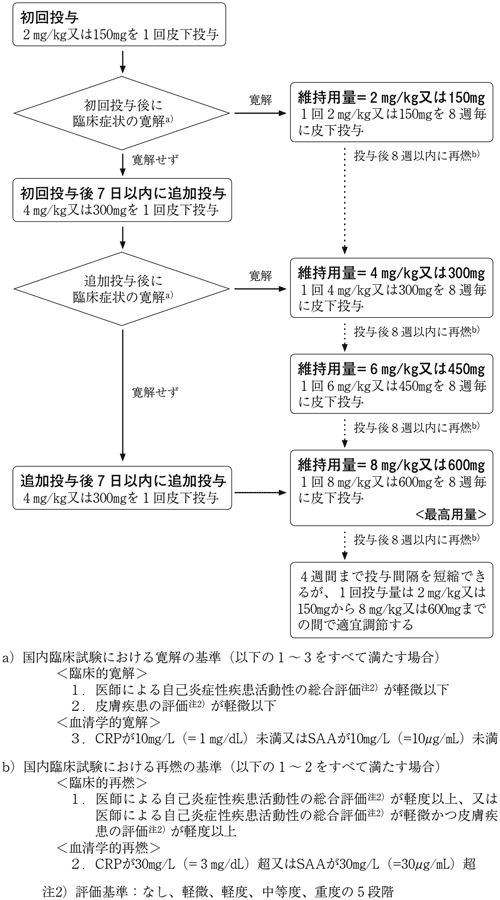
-
〈高IgD症候群(メバロン酸キナーゼ欠損症)〉
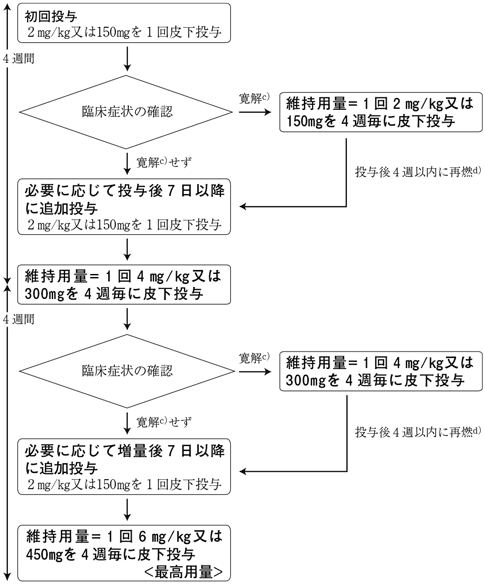
-
〈TNF受容体関連周期性症候群及び家族性地中海熱〉
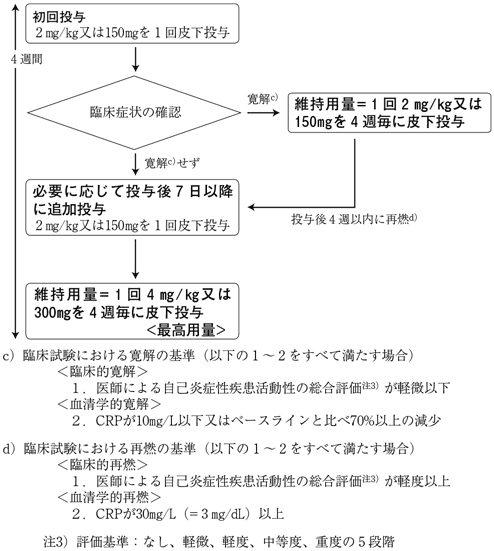
-
**〈シュニッツラー症候群〉
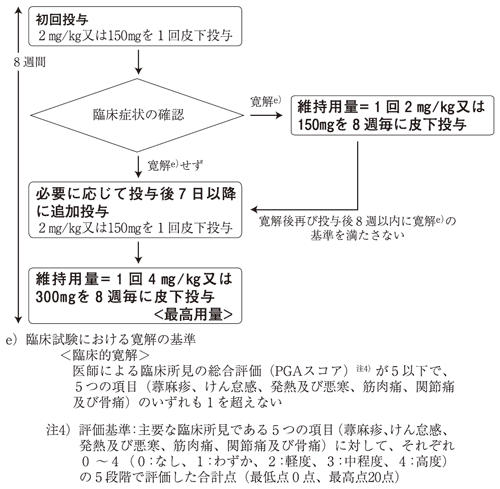
8. 重要な基本的注意
- 8.1 臨床試験において、上気道感染等の感染症が高頻度に報告されており、重篤な感染症も報告されているため、本剤投与中は感染症の発現、再発及び増悪に十分注意すること。[1.1 参照],[1.2 参照],[2.1 参照],[9.1.1 参照],[9.1.3 参照],[9.1.4 参照],[11.1.1 参照]
- 8.2 本剤により感染に対する炎症反応が抑制される可能性があるため、本剤投与中は患者の状態を十分に観察すること。[1.1 参照],[1.2 参照],[2.1 参照],[9.1.1 参照],[9.1.3 参照],[9.1.4 参照],[11.1.1 参照]
- 8.3 本剤投与に先立って結核に関する十分な問診及び胸部X線(レントゲン)検査に加えインターフェロンγ遊離試験又はツベルクリン反応検査を行い、適宜胸部CT検査等を行うことにより、結核感染の有無を確認すること。また、本剤投与中も、胸部X線検査等の適切な検査を定期的に行うなど結核の発現には十分に注意し、患者に対し、結核を疑う症状(持続する咳、体重減少、発熱等)が発現した場合には速やかに担当医に連絡するよう説明すること。なお、結核の活動性が確認された場合は結核の治療を優先し、本剤を投与しないこと。[2.2 参照],[9.1.2 参照]
- 8.4 本剤投与により好中球減少があらわれることがあるので、初回投与前、概ね投与1ヵ月後、及びその後本剤投与中は定期的に好中球数を測定すること。[11.1.2 参照]
- 8.5 臨床試験において、アナフィラキシー又はアナフィラキシーショックは報告されていないが、本剤の投与に対する過敏症反応が報告されているため、重篤な過敏症反応のリスクを除外することはできない。本剤を投与する際には過敏症反応の発現に注意し、必要に応じて適切な処置を行うこと。
- 8.6 本剤を投与された患者において、悪性腫瘍が報告されている。本剤を含む抗IL-1製剤との関連性は明らかではないが、悪性腫瘍等の発現には注意すること。[1.1 参照]
- 8.7 本剤投与中は、生ワクチン接種による感染症発現のリスクを否定できないため、生ワクチン接種は行わないこと。本剤投与前に、必要なワクチンを接種しておくことが望ましい。
- 8.8 抗リウマチ生物製剤によるB型肝炎ウイルスの再活性化が報告されているので、本剤投与に先立って、B型肝炎ウイルス感染の有無を確認すること。[9.1.5 参照]
- 8.9 他の生物製剤から変更する場合は、感染症の徴候について患者の状態を十分に観察すること。
- 8.10 本剤は、マスターセルバンク作製時において、培地成分の一部としてヒト血清アルブミン及びヒト血清トランスフェリンを使用しているが、最終製品の成分としては含まれていない。これらヒト血液由来成分のうち、ヒト血清アルブミンの原血漿に対してC型肝炎ウイルス(HCV)に対する核酸増幅検査を実施している。原血漿を対象としたその他の核酸増幅検査は実施していないが、血清学的検査によりウイルスの抗原又はウイルスに対する抗体が陰性であることを確認している。更に、これらヒト血液由来成分及びカナキヌマブ(遺伝子組換え)の製造において、複数の工程によりウイルスの除去・不活化をしており、最終製品へのB型肝炎ウイルス(HBV)、C型肝炎ウイルス(HCV)及びヒト免疫不全ウイルス(HIV-1及びHIV-2)混入の可能性は極めて低い。また、ヒト血清アルブミンの製造にオランダで採血したヒト血液を用いているが、本剤の投与により伝達性海綿状脳症(TSE)がヒトに伝播したとの報告はなく、TSEに関する理論的なリスク評価値は、一定の安全性を確保する目安に達しており、本剤によるTSE伝播のリスクは極めて低い。本剤の投与に際しては、その旨の患者又はその保護者への説明を考慮すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
-
9.1.1 感染症(重篤な感染症を除く)の患者又は感染症が疑われる患者
感染症が悪化するおそれがある。[1.1 参照],[1.2 参照],[2.1 参照],[8.1 参照],[8.2 参照],[11.1.1 参照]
-
9.1.2 結核の既往歴を有する患者又は結核感染が疑われる患者
結核の診療経験がある医師に相談すること。結核を活動化させるおそれがある。以下のいずれかの患者には、原則として抗結核薬を投与した上で、本剤を投与すること。
-
9.1.3 再発性感染症の既往歴のある患者
感染症が再発するおそれがある。[1.1 参照],[1.2 参照],[2.1 参照],[8.1 参照],[8.2 参照],[11.1.1 参照]
-
9.1.4 易感染性の状態にある患者
感染症を誘発するおそれがある。[1.1 参照],[1.2 参照],[2.1 参照],[8.1 参照],[8.2 参照],[11.1.1 参照]
-
9.1.5 B型肝炎ウイルスキャリアの患者又は既往感染者(HBs抗原陰性、かつHBc抗体又はHBs抗体陽性)
最新のB型肝炎治療ガイドラインを参考に肝機能検査値や肝炎ウイルスマーカーのモニタリングを行うなど、B型肝炎ウイルスの再活性化の徴候や症状の発現に注意すること。[8.8 参照]
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。動物実験(マーモセット)で胎児への移行が認められている。
9.6 授乳婦
9.7 小児等
低出生体重児、新生児、乳児又は2歳未満の幼児に対する安全性及び有効性を検討することを目的とした臨床試験は実施していない。
9.8 高齢者
一般に生理機能が低下しているので注意すること。
10. 相互作用
- 本剤と他の薬剤との相互作用を検討した臨床試験は実施されていない。
代謝酵素チトクロームP450(CYP450)の発現は、IL-1β等の炎症性サイトカインにより抑制されているとの報告があり、本剤のIL-1β阻害作用により、CYP450の発現が増加する可能性がある。CYP450により代謝され、治療域が狭い薬剤と併用する場合には、これらの薬剤の効果や血中濃度に関するモニタリングを行い、必要に応じて投与量を調節すること。
10.2 併用注意(併用に注意すること)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
抗TNF製剤 |
重篤な感染症発現のリスクが増大するおそれがある。また、他の抗IL-1製剤と抗TNF製剤との併用により、重篤な感染症の発現頻度増加が認められているため、本剤との併用は行わないことが望ましい。 |
共に免疫抑制作用を有するため。 |
11. 副作用
11.1 重大な副作用
-
11.1.1 **,*重篤な感染症(11.8%)
敗血症や日和見感染症(アスペルギルス症、非定型抗酸菌症、帯状疱疹等)等の重篤な感染症があらわれることがある。[1.1 参照],[1.2 参照],[2.1 参照],[8.1 参照],[8.2 参照],[9.1.1 参照],[9.1.3 参照],[9.1.4 参照]
- 11.1.2 好中球減少(頻度不明)
11.2 その他の副作用
5%以上 |
5%未満 |
頻度不明 |
|
|---|---|---|---|
*感染症 |
鼻咽頭炎 |
胃腸炎、肺炎、副鼻腔炎、上気道感染、咽頭炎、尿路感染 |
気管支炎、ウイルス感染、扁桃炎、鼻炎、耳感染、外陰部膣カンジダ症、下気道感染、肺感染 |
神経系 |
- |
頭痛 |
回転性めまい |
*過敏症 |
過敏症反応 |
- |
- |
皮膚 |
注射部位反応 |
- |
- |
*消化器 |
- |
口内炎、下痢 |
腹痛 |
肝臓 |
- |
AST・ALT上昇 |
- |
血液 |
- |
白血球数減少 |
血小板数減少 |
その他 |
- |
- |
体重増加 |
14. 適用上の注意
14.1 薬剤調製時の注意
14.2 薬剤投与時の注意
- 14.2.1 溶液内に粒子がある場合等、外観に異常を認めた場合には使用しないこと。
- 14.2.2 バイアルのゴム栓部分をアルコール綿等で消毒する。
- 14.2.3 投与量に応じて必要な液量を、21ゲージの注射針を装着した注射筒を用いて注意深く採取する。このとき、必要液量を正確に採取できる注射筒を用いること。
- 14.2.4 採取後、27ゲージの注射針を用いて皮下投与する。
- 14.2.5 瘢痕組織への投与を避けること。
- 14.2.6 1回につき1.0mLを超えて投与する場合には、1箇所あたり1.0mLを超えないように部位を分けて投与すること。
- 14.2.7 1バイアルは1回のみの使用とし、使用後の残液は微生物汚染のおそれがあるので、再使用しないこと。
1. 警告
- 1.1 本剤投与により、敗血症を含む重篤な感染症等があらわれることがあり、本剤との関連性は明らかではないが、悪性腫瘍の発現も報告されている。本剤が疾病を完治させる薬剤でないことも含め、これらの情報を患者に十分説明し、患者が理解したことを確認した上で、治療上の有益性が危険性を上回ると判断される場合にのみ本剤を投与すること。また、本剤の投与において、重篤な感染症等の副作用により、致命的な経過をたどることがあるので、緊急時に十分に措置できる医療施設及び医師のもとで投与し、本剤投与後に副作用が発現した場合には、速やかに担当医に連絡するよう患者に注意を与えること。[1.2 参照],[2.1 参照],[8.1 参照],[8.2 参照],[8.6 参照],[9.1.1 参照],[9.1.3 参照],[9.1.4 参照],[11.1.1 参照]
- 1.2 敗血症等の致命的な感染症が報告されているため、十分な観察を行うなど感染症の発現に注意すること。[1.1 参照],[2.1 参照],[8.1 参照],[8.2 参照],[9.1.1 参照],[9.1.3 参照],[9.1.4 参照],[11.1.1 参照]
- 1.3 本剤についての十分な知識と適応疾患の治療の知識・経験をもつ医師が使用すること。
2. 禁忌(次の患者には投与しないこと)
- 2.1 重篤な感染症の患者[感染症が悪化するおそれがある。][1.1 参照],[1.2 参照],[8.1 参照],[8.2 参照],[9.1.1 参照],[9.1.3 参照],[9.1.4 参照],[11.1.1 参照]
- 2.2 活動性結核の患者[症状が悪化するおそれがある。][8.3 参照],[9.1.2 参照]
- 2.3 本剤の成分に対し過敏症の既往歴のある患者
5. 効能又は効果に関連する注意
-
**〈シュニッツラー症候群〉
- 5.1 「17. 臨床成績」の項の内容を熟知し、臨床試験に組み入れられた患者の臨床症状及び治療歴を十分に理解した上で、適応患者を選択すること。[17.1.6 参照]
- 〈家族性地中海熱〉
- *〈全身型若年性特発性関節炎及び成人発症スチル病〉
6. 用法及び用量
-
〈クリオピリン関連周期性症候群〉
通常、体重40kg以下の患者にはカナキヌマブ(遺伝子組換え)として1回2mg/kgを、体重40kgを超える患者には1回150mgを8週毎に皮下投与する。
十分な臨床的効果(皮疹及び炎症症状の寛解)がみられない場合には適宜漸増するが、1回最高用量は体重40kg以下の患者では8mg/kg、体重40kgを超える患者では600mgとする。
最高用量まで増量し、8週以内に再燃がみられた場合には、投与間隔を4週間まで短縮できる。
なお、症状に応じて1回投与量の増減を検討すること。 -
〈高IgD症候群(メバロン酸キナーゼ欠損症)〉
通常、体重40kg以下の患者にはカナキヌマブ(遺伝子組換え)として1回2mg/kgを、体重40kgを超える患者には1回150mgを、4週毎に皮下投与する。
十分な臨床的効果がみられない場合には追加投与又は適宜漸増するが、1回最高用量は体重40kg以下の患者では6mg/kg、体重40kgを超える患者では450mgとする。 -
〈TNF受容体関連周期性症候群及び家族性地中海熱〉
通常、体重40kg以下の患者にはカナキヌマブ(遺伝子組換え)として1回2mg/kgを、体重40kgを超える患者には1回150mgを、4週毎に皮下投与する。
十分な臨床的効果がみられない場合には追加投与又は適宜漸増するが、1回最高用量は体重40kg以下の患者では4mg/kg、体重40kgを超える患者では300mgとする。 -
〈シュニッツラー症候群〉
**通常、成人にはカナキヌマブ(遺伝子組換え)として体重40kg以下の患者では1回2mg/kgを、体重40kgを超える患者では1回150mgを8週毎に皮下投与する。
十分な臨床的効果がみられない場合には追加投与又は適宜漸増するが、1回最高用量は体重40kg以下の患者では4mg/kg、体重40kgを超える患者では300mgとする。 -
*〈全身型若年性特発性関節炎及び成人発症スチル病〉
通常、カナキヌマブ(遺伝子組換え)として1回4mg/kgを、4週毎に皮下投与する。1回最高用量は300mgとする。
7. 用法及び用量に関連する注意
- 〈効能共通〉
-
**〈クリオピリン関連周期性症候群、高IgD症候群(メバロン酸キナーゼ欠損症)、TNF受容体関連周期性症候群、シュニッツラー症候群、家族性地中海熱〉
- 7.3 投与は1回2mg/kg又は150mgの低用量から開始し、十分な効果がみられない、もしくは再燃がみられた場合に限り、下図を参考に投与量の増量を行うこと。[17.1.1 参照],[17.1.5 参照],[17.1.6 参照]
- 十分な臨床的効果がみられない場合の漸増方法
-
〈クリオピリン関連周期性症候群〉
-
〈高IgD症候群(メバロン酸キナーゼ欠損症)〉
-
〈TNF受容体関連周期性症候群及び家族性地中海熱〉
-
**〈シュニッツラー症候群〉
8. 重要な基本的注意
- 8.1 臨床試験において、上気道感染等の感染症が高頻度に報告されており、重篤な感染症も報告されているため、本剤投与中は感染症の発現、再発及び増悪に十分注意すること。[1.1 参照],[1.2 参照],[2.1 参照],[9.1.1 参照],[9.1.3 参照],[9.1.4 参照],[11.1.1 参照]
- 8.2 本剤により感染に対する炎症反応が抑制される可能性があるため、本剤投与中は患者の状態を十分に観察すること。[1.1 参照],[1.2 参照],[2.1 参照],[9.1.1 参照],[9.1.3 参照],[9.1.4 参照],[11.1.1 参照]
- 8.3 本剤投与に先立って結核に関する十分な問診及び胸部X線(レントゲン)検査に加えインターフェロンγ遊離試験又はツベルクリン反応検査を行い、適宜胸部CT検査等を行うことにより、結核感染の有無を確認すること。また、本剤投与中も、胸部X線検査等の適切な検査を定期的に行うなど結核の発現には十分に注意し、患者に対し、結核を疑う症状(持続する咳、体重減少、発熱等)が発現した場合には速やかに担当医に連絡するよう説明すること。なお、結核の活動性が確認された場合は結核の治療を優先し、本剤を投与しないこと。[2.2 参照],[9.1.2 参照]
- 8.4 本剤投与により好中球減少があらわれることがあるので、初回投与前、概ね投与1ヵ月後、及びその後本剤投与中は定期的に好中球数を測定すること。[11.1.2 参照]
- 8.5 臨床試験において、アナフィラキシー又はアナフィラキシーショックは報告されていないが、本剤の投与に対する過敏症反応が報告されているため、重篤な過敏症反応のリスクを除外することはできない。本剤を投与する際には過敏症反応の発現に注意し、必要に応じて適切な処置を行うこと。
- 8.6 本剤を投与された患者において、悪性腫瘍が報告されている。本剤を含む抗IL-1製剤との関連性は明らかではないが、悪性腫瘍等の発現には注意すること。[1.1 参照]
- 8.7 本剤投与中は、生ワクチン接種による感染症発現のリスクを否定できないため、生ワクチン接種は行わないこと。本剤投与前に、必要なワクチンを接種しておくことが望ましい。
- 8.8 抗リウマチ生物製剤によるB型肝炎ウイルスの再活性化が報告されているので、本剤投与に先立って、B型肝炎ウイルス感染の有無を確認すること。[9.1.5 参照]
- 8.9 他の生物製剤から変更する場合は、感染症の徴候について患者の状態を十分に観察すること。
- 8.10 本剤は、マスターセルバンク作製時において、培地成分の一部としてヒト血清アルブミン及びヒト血清トランスフェリンを使用しているが、最終製品の成分としては含まれていない。これらヒト血液由来成分のうち、ヒト血清アルブミンの原血漿に対してC型肝炎ウイルス(HCV)に対する核酸増幅検査を実施している。原血漿を対象としたその他の核酸増幅検査は実施していないが、血清学的検査によりウイルスの抗原又はウイルスに対する抗体が陰性であることを確認している。更に、これらヒト血液由来成分及びカナキヌマブ(遺伝子組換え)の製造において、複数の工程によりウイルスの除去・不活化をしており、最終製品へのB型肝炎ウイルス(HBV)、C型肝炎ウイルス(HCV)及びヒト免疫不全ウイルス(HIV-1及びHIV-2)混入の可能性は極めて低い。また、ヒト血清アルブミンの製造にオランダで採血したヒト血液を用いているが、本剤の投与により伝達性海綿状脳症(TSE)がヒトに伝播したとの報告はなく、TSEに関する理論的なリスク評価値は、一定の安全性を確保する目安に達しており、本剤によるTSE伝播のリスクは極めて低い。本剤の投与に際しては、その旨の患者又はその保護者への説明を考慮すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
-
9.1.1 感染症(重篤な感染症を除く)の患者又は感染症が疑われる患者
感染症が悪化するおそれがある。[1.1 参照],[1.2 参照],[2.1 参照],[8.1 参照],[8.2 参照],[11.1.1 参照]
-
9.1.2 結核の既往歴を有する患者又は結核感染が疑われる患者
結核の診療経験がある医師に相談すること。結核を活動化させるおそれがある。以下のいずれかの患者には、原則として抗結核薬を投与した上で、本剤を投与すること。
-
9.1.3 再発性感染症の既往歴のある患者
感染症が再発するおそれがある。[1.1 参照],[1.2 参照],[2.1 参照],[8.1 参照],[8.2 参照],[11.1.1 参照]
-
9.1.4 易感染性の状態にある患者
感染症を誘発するおそれがある。[1.1 参照],[1.2 参照],[2.1 参照],[8.1 参照],[8.2 参照],[11.1.1 参照]
-
9.1.5 B型肝炎ウイルスキャリアの患者又は既往感染者(HBs抗原陰性、かつHBc抗体又はHBs抗体陽性)
最新のB型肝炎治療ガイドラインを参考に肝機能検査値や肝炎ウイルスマーカーのモニタリングを行うなど、B型肝炎ウイルスの再活性化の徴候や症状の発現に注意すること。[8.8 参照]
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。動物実験(マーモセット)で胎児への移行が認められている。
9.6 授乳婦
9.7 小児等
低出生体重児、新生児、乳児又は2歳未満の幼児に対する安全性及び有効性を検討することを目的とした臨床試験は実施していない。
9.8 高齢者
一般に生理機能が低下しているので注意すること。
10. 相互作用
- 本剤と他の薬剤との相互作用を検討した臨床試験は実施されていない。
代謝酵素チトクロームP450(CYP450)の発現は、IL-1β等の炎症性サイトカインにより抑制されているとの報告があり、本剤のIL-1β阻害作用により、CYP450の発現が増加する可能性がある。CYP450により代謝され、治療域が狭い薬剤と併用する場合には、これらの薬剤の効果や血中濃度に関するモニタリングを行い、必要に応じて投与量を調節すること。
10.2 併用注意(併用に注意すること)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
抗TNF製剤 |
重篤な感染症発現のリスクが増大するおそれがある。また、他の抗IL-1製剤と抗TNF製剤との併用により、重篤な感染症の発現頻度増加が認められているため、本剤との併用は行わないことが望ましい。 |
共に免疫抑制作用を有するため。 |
11. 副作用
11.1 重大な副作用
-
11.1.1 **,*重篤な感染症(11.8%)
敗血症や日和見感染症(アスペルギルス症、非定型抗酸菌症、帯状疱疹等)等の重篤な感染症があらわれることがある。[1.1 参照],[1.2 参照],[2.1 参照],[8.1 参照],[8.2 参照],[9.1.1 参照],[9.1.3 参照],[9.1.4 参照]
- 11.1.2 好中球減少(頻度不明)
11.2 その他の副作用
5%以上 |
5%未満 |
頻度不明 |
|
|---|---|---|---|
*感染症 |
鼻咽頭炎 |
胃腸炎、肺炎、副鼻腔炎、上気道感染、咽頭炎、尿路感染 |
気管支炎、ウイルス感染、扁桃炎、鼻炎、耳感染、外陰部膣カンジダ症、下気道感染、肺感染 |
神経系 |
- |
頭痛 |
回転性めまい |
*過敏症 |
過敏症反応 |
- |
- |
皮膚 |
注射部位反応 |
- |
- |
*消化器 |
- |
口内炎、下痢 |
腹痛 |
肝臓 |
- |
AST・ALT上昇 |
- |
血液 |
- |
白血球数減少 |
血小板数減少 |
その他 |
- |
- |
体重増加 |
14. 適用上の注意
14.1 薬剤調製時の注意
14.2 薬剤投与時の注意
- 14.2.1 溶液内に粒子がある場合等、外観に異常を認めた場合には使用しないこと。
- 14.2.2 バイアルのゴム栓部分をアルコール綿等で消毒する。
- 14.2.3 投与量に応じて必要な液量を、21ゲージの注射針を装着した注射筒を用いて注意深く採取する。このとき、必要液量を正確に採取できる注射筒を用いること。
- 14.2.4 採取後、27ゲージの注射針を用いて皮下投与する。
- 14.2.5 瘢痕組織への投与を避けること。
- 14.2.6 1回につき1.0mLを超えて投与する場合には、1箇所あたり1.0mLを超えないように部位を分けて投与すること。
- 14.2.7 1バイアルは1回のみの使用とし、使用後の残液は微生物汚染のおそれがあるので、再使用しないこと。